Aciclovir
 |
|
|---|---|
 |
|
| Systematic (IUPAC) name | |
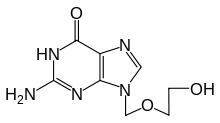
| 2-amino-9-((2-hydroxyethoxy)methyl)-1H-purin-6(9H)-one | |
| Identifiers | |
| CAS number | 59277-89-3 |
| ATC code | J05AB01 D06BB03 S01AD03 |
| PubChem | CID 2022 |
| DrugBank | APRD00567 |
| ChemSpider | 1945 |
| Chemical data | |
| Formula | C8H11N5O3 |
| Mol. mass | 225.21 g/mol |
| SMILES | eMolecules & PubChem |
| Synonyms | acycloguanosine |
| Physical data | |
| Melt. point | 256.5 °C (494 °F) |
| Pharmacokinetic data | |
| Bioavailability | 10–20% (oral) |
| Protein binding | 9–33% |
| Metabolism | Viral thymidine kinase |
| Half-life | 2.2–20 hours |
| Excretion | Renal |
| Therapeutic considerations | |
| Pregnancy cat. | B3 (Au), B (U.S.) |
| Legal status | unscheduled/S4 (Au), POM (UK) |
| Routes | Intravenous, oral, topical |
| |
|
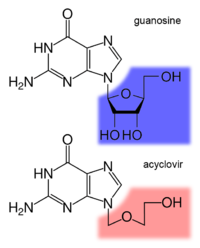
Aciclovir (INN) (pronounced /eɪˈsaɪklɵvɪər/) or acyclovir (USAN, former BAN), chemical name acycloguanosine, abbreviated as ACV,[1] is a guanosine analogue antiviral drug, marketed under trade names such as Cyclovir, Herpex, Acivir, Acivirax(Mash-Premier), Zovirax, Aciclovir (Sanofi-Aventis) and Zovir (GSK). One of the most commonly-used antiviral drugs, it is primarily used for the treatment of herpes simplex virus infections, as well as in the treatment of herpes zoster (shingles).
Aciclovir was seen as the start of a new era in antiviral therapy,[1] as it is extremely selective and low in cytotoxicity. Pharmacologist Gertrude B. Elion was awarded the 1988 Nobel Prize in Medicine, partly for the development of aciclovir. Dr. Richard Whitley, a University of Alabama at Birmingham researcher and pioneer in antiviral therapy, was the first to successfully use the drug in humans.
Contents |
Pharmacology
Mechanism of action
Aciclovir differs from previous nucleoside analogues in that it contains only a partial nucleoside structure: the sugar ring is replaced by an open-chain structure. It is selectively converted into acyclo-guanosine monophosphate (acyclo-GMP) by viral thymidine kinase, which is far more effective (3000 times) in phosphorylation than cellular thymidine kinase. Subsequently, the monophosphate form is further phosphorylated into the active triphosphate form, acyclo-guanosine triphosphate (acyclo-GTP), by cellular kinases. Acyclo-GTP is a very potent inhibitor of viral DNA polymerase; it has approximately 100 times greater affinity for viral than cellular polymerase. As a substrate, acyclo-GTP is incorporated into viral DNA, resulting in chain termination. It has also been shown that viral enzymes cannot remove acyclo-GTP from the chain, which results in inhibition of further activity of DNA polymerase. Acyclo-GTP is fairly rapidly metabolised within the cell, possibly by cellular phosphatases.
In sum, aciclovir can be considered a prodrug: it is administered in an inactive (or less active) form and is metabolised into a more active species after administration.
Microbiology
Aciclovir is active against most known species in the herpesvirus family. In descending order of activity:[2]
- Herpes simplex virus type I (HSV-1)
- Herpes simplex virus type II (HSV-2)
- Varicella zoster virus (VZV)
- Epstein-Barr virus (EBV)
- Cytomegalovirus (CMV) -- least activity
Activity is predominantly against HSV, and to a lesser extent VZV. It is only of limited efficacy against EBV and CMV. It is inactive against latent viruses in nerve ganglia.
Resistance
To date, resistance to aciclovir has not been clinically significant. Mechanisms of resistance in HSV include deficient viral thymidine kinase; and mutations to viral thymidine kinase and/or DNA polymerase, altering substrate sensitivity.[3] Acyclovir has also shown cross-resistance with Valacyclovir and Famcyclovir.
Pharmacokinetics
Aciclovir is poorly water soluble and has poor oral bioavailability (15-30%), hence intravenous administration is necessary if high concentrations are required. When orally administered, peak plasma concentration occurs after 1–2 hours. Aciclovir has a high distribution rate, only 30% is protein-bound in plasma. The elimination half-life of aciclovir is approximately 3 hours. It is renally excreted, partly by glomerular filtration and partly by tubular secretion.
The poor oral bioavailability may also be improved by administering Valaciclovir, which has an oral bioavailability of about 55%. Valaciclovir is then converted to Aciclovir by esterases via hepatic first-pass metabolism.
Clinical use

Indications
Aciclovir is indicated for the treatment of HSV and VZV infections, including:[4]
- Genital herpes simplex (treatment and prophylaxis)
- Herpes simplex labialis (cold sores)
- Herpes zoster (shingles)
- Acute chickenpox in immunocompromised patients
- Herpes simplex encephalitis
- Acute mucocutaneous HSV infections in immunocompromised patients
- Herpes simplex keratitis (ocular herpes)
- Herpes simplex blepharitis (not to be mistaken with ocular herpes)
- Bell's Palsy
HIV-1 progression can be delayed by using Aciclovir, according to study lead by Dr Jairam Lingappa. Effective in 16% of cases, can delay the HAART treatment by 1–2 years. University of Washington, Seattle. During a 2 year trial, 284 people progressed with the HIV-1, versus 324 who had not been treated with Aciclovir. [5] It has been claimed that the evidence for the effectiveness of topically applied cream for recurrent labial outbreaks is weak.[6] An earlier review of scientific literature showed that there is some effect in reducing the number and duration of lesions if aciclovir is applied at an early stage of an outbreak.[7] However, it was concluded that oral therapy for episodes is inappropriate for most non-immunocompromised patients based on costs and benefits, presumably in countries where aciclovir is only available on prescription. It was concluded that there is evidence for an oral prophylactic role in preventing recurrences.
Dosage forms
Aciclovir is commonly marketed as tablets (200 mg, 400 mg, 800 mg and 1 gram), topical cream (5%), intravenous injection (25 mg/mL) and ophthalmic ointment (3%). Cream preparations are used primarily for labial herpes simplex. The intravenous injection is used when high concentrations of aciclovir are required. The ophthalmic ointment preparation is only used for herpes simplex keratitis. In Singapore, it is available as a 400 mg preparation known as Avorax.
Adverse effects
Systemic therapy
Common adverse drug reactions (≥1% of patients) associated with systemic acyclovir therapy (oral or IV) include: nausea, vomiting, diarrhea and/or headache. In high doses, hallucinations have been reported. Infrequent adverse effects (0.1–1% of patients) include: agitation, vertigo, confusion, dizziness, oedema, arthralgia, sore throat, constipation, abdominal pain,hair loss, rash and/or weakness. Rare adverse effects (<0.1% of patients) include: coma, seizures, neutropenia, leukopenia, crystalluria, anorexia, fatigue, hepatitis, Stevens-Johnson syndrome, toxic epidermal necrolysis and/or anaphylaxis.[4]
Additional common adverse effects, when acyclovir is administered IV, include encephalopathy (1% of patients) and injection site reactions. The injection formulation is alkaline (pH 11), and extravasation may cause local tissue pain and irritation.[4] Renal impairment has been reported when acyclovir is given in large, fast doses intravenously, due to the crystallisation of acyclovir in the kidneys.[8][9]
Topical therapy
Acyclovir topical cream is commonly associated (≥1% of patients) with: dry or flaking skin or transient stinging/burning sensations. Infrequent adverse effects include erythema or itch.[4]
When applied to the eye, acyclovir is commonly associated (≥1% of patients) with transient mild stinging. Infrequently (0.1–1% of patients), ophthalmic aciclovir is associated with superficial punctate keratitis or allergic reactions.[4]
Toxicity
Since acyclovir can also be incorporated into cellular DNA, it is a chromosome mutagen, therefore, its use should be avoided during pregnancy. However it has not been shown to cause any teratogenic or carcinogenic effects and is frequently prescribed for pregnant women. The acute toxicity (LD50) of acyclovir when given orally is greater than 1 g/kg, due to its low oral bioavailability. Patients with renal impairment often exhibit elimination half-lives for the drug that are 5-6 times longer than in those with normal renal function, leading to accumulation of acyclovir in the plasma and the likelihood of development of toxic reactions such as lethargy, confusion and myoclonus.[10]
Detection in biological fluids
Acyclovir may be quantitated in plasma or serum in order to monitor for drug accumulation in patients with renal dysfunction or to confirm a diagnosis of poisoning in acute overdose victims.[10]
See also
- Valaciclovir
References
- ↑ 1.0 1.1 de Clercq, Erik; Field, Hugh J (5 October 2005). "Antiviral prodrugs — the development of successful prodrug strategies for antiviral chemotherapy". British Journal of Pharmacology (Wiley-Blackwell) 147 (1): pp. 1–11. January 2006. doi:10.1038/sj.bjp.0706446. PMID 16284630
- ↑ O'Brien JJ, Campoli-Richards DM. (1989). "Aciclovir. An updated review of its antiviral activity, pharmacokinetic properties and therapeutic efficacy". Drugs 37 (3): 233–309. PMID 2653790.
- ↑ Sweetman S, editor. Martindale: The complete drug reference. 34th ed. London: Pharmaceutical Press; 2004. ISBN 0-85369-550-4
- ↑ 4.0 4.1 4.2 4.3 4.4 Rossi S, editor. Australian Medicines Handbook 2006. Adelaide: Australian Medicines Handbook; 2006. ISBN 0-9757919-2-3
- ↑ "HIV illness 'delayed by' herpes drug aciclovir". http://news.bbc.co.uk/2/hi/health/8512412.stm. Retrieved 20. May 2010.
- ↑ Graham Worrall (6 Jul 1996). "Evidence for efficacy of topical aciclovir in recurrent herpes labialis is weak". BMJ 313: 46. http://www.bmj.com/cgi/content/full/313/7048/46/a. - Letter
- ↑ Graham Worrall (6 Jan 1996). "Acyclovir in recurrent herpes labialis". BMJ 312 (7022): 6. PMID 8555890. PMC 2349724. http://www.bmj.com/cgi/content/full/312/7022/6. - Editorial
- ↑ Brigden D, Rosling AE, Woods NC (July 1982). "Renal function after acyclovir intravenous injection". The American Journal of Medicine 73 (1A): 182–5. doi:10.1016/0002-9343(82)90087-0. PMID 6285711.
- ↑ Sawyer MH, Webb DE, Balow JE, Straus SE (June 1988). "Acyclovir-induced renal failure. Clinical course and histology". The American Journal of Medicine 84 (6): 1067–71. doi:10.1016/0002-9343(88)90313-0. PMID 3376977.
- ↑ 10.0 10.1 R. Baselt, Disposition of Toxic Drugs and Chemicals in Man, 8th edition, Biomedical Publications, Foster City, CA, 2008, pp. 29-31.
Further reading
- Harvey Stewart C. in Remington’s Pharmaceutical Sciences 18th edition: (ed. Gennard, Alfonso R.) Mack Publishing Company, 1990. ISBN 0-912734-04-3.
- Huovinen P., Valtonen V. in Kliininen Farmakologia (ed. Neuvonen et al.). Kandidaattikustannus Oy, 1994. ISBN 951-8951-09-8.
- Périgaud C., Gosselin G., Imbach J. -L.: Nucleoside analogues as chemotherapeutic agents: a review. Nucleosides and nucleotides 1992; 11(2-4)
- Rang H.P., Dale M.M., Ritter J.M.: Pharmacology, 3rd edition. Pearson Professional Ltd, 1995. 2003 (5th) edition ISBN 0-443-07145-4; 2001 (4th) edition ISBN 0-443-06574-8; 1990 edition ISBN 0-443-03407-9.
External links
|
||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||