Adrenoleukodystrophy
| Adrenoleukodystrophy | |
|---|---|
| Classification and external resources | |
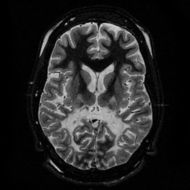 T2 weighted axial scan at the level of the caudate heads demonstrates marked loss of posterior white matter, with reduced volume and increased signal intensity. The anterior white matter is spared. Features are consistent with X-linked adrenoleukodystrophy. |
|
| ICD-10 | E71.3 |
| ICD-9 | 330.0, 277.86 |
| OMIM | 300100 202370 |
| DiseasesDB | 292 |
| MeSH | D000326 |
Adrenoleukodystrophy (ALD), also called "Siemerling-Creutzfeldt Disease," or "Schilder's disease"[1]:545) is a rare, inherited disorder that leads to progressive brain damage, failure of the adrenal glands and eventually death. ALD is a disease in a group of genetic disorders called leukodystrophies. Adrenoleukodystrophy progressively damages the myelin sheath, a complex fatty neural tissue that insulates many nerves of the central and peripheral nervous systems, eventually destroying it. Without myelin, nerves are unable to conduct an impulse, leading to increasing disability as myelin destruction increases and intensifies.
An essential protein, called a transporter protein, is missing in ALD patients. This protein is needed to carry an enzyme which is used to break down very long-chain fatty acids found in the normal diet. Lack of this protein can give rise to a build-up of very long-chain fatty acids (VLCFA), in the body which can damage the brain and the adrenal gland.
There are several different types of the disease which can be inherited, but the most common form is an X-linked condition. Patients with X-linked ALD are all male, but about one in five women with the disease gene develop some symptoms. Adrenomyeloneuropathy is a less-severe form of ALD, with onset of symptoms occurring in adolescence or adulthood. This milder form does not include cerebral involvement, and should be included in the differential diagnosis of all males with adrenal insufficiency.
Although this disorder affects the growth and/or development of myelin, leukodystrophies are different from demyelinating disorders such as multiple sclerosis where myelin is formed normally but is lost by immunologic dysfunction or for other reasons.
Contents |
Symptoms
The clinical presentation is largely dependent on the age of onset of the disease. The classical, severe type is the childhood cerebral form which, as an X-linked disease, affects males. Symptoms normally start between the ages of 4 and 10 and include loss of previously acquired neurologic abilities, seizures, ataxia, Addison's disease, and degeneration of visual and auditory function. It has been seen that infants that have been positively diagnosed by the age of 1 year old have usually become very ill by the age of 10 to 12 years and die soon after. This severe form of the disease was first described by Ernst Siemerling and Hans Gerhard Creutzfeldt.[2] A similar form can also occur in adolescents and very rarely in adults. Addison's disease can be an initial symptom of ALD, and many pediatric endocrinologists will measure very long chain free fatty acids in newly diagnosed males with this condition, as a screening test for ALD.
In another form of ALD, one primarily strikes young men, the spinal cord dysfunction is more prominent and is therefore called adrenomyeloneuropathy, or "AMN." The patients usually present with weakness and numbness of the limbs and urination or defecation problems. Most victims of this form are also males, although some female carriers exhibit symptoms of AMN. [3]
Adult and neonatal forms of the disease also exist but they are extremely rare. (These tend to affect both males and females and be inherited in an autosomal recessive manner.) Some patients may present with sole findings of adrenal insufficiency. ALD also causes uncontrollable rage in some cases.
Diagnosis
The diagnosis is established by clinical findings and the detection of serum very long-chain free fatty acid levels.[4] MRI examination reveals white matter abnormalities, and neuro-imaging findings of this disease are somewhat reminiscent of the findings of multiple sclerosis. Genetic testing for the analysis of the defective gene is available in some centers.
Neonatal screening may become available in the future, which may permit early diagnosis and treatment.[5] Approximately 1 in 42,000 boys are diagnosed with X-linked ALD. [6]
Genetics
X-linked
The most common form of ALD is X-linked, which means that the defective gene is on the X chromosome. It is located at Xq28, and the disease is characterized by excessive accumulation of very long-chain fatty acids (VLCFA), which are fatty acids with chains of 25–30 carbon atoms. The most common is hexacosanoate, with a 26 carbon skeleton. The elevation in (VLCFA) was originally described by Moser et al. in 1981.[7] The ALD gene was discovered in 1993, and it coded for a protein that was a member of a family of transporter proteins, not an enzyme. It is unknown how high levels of very long chain fatty acids cause the loss of myelin.
The gene (ABCD1 or "ATP-binding cassette, subfamily D, member 1") codes for a protein that transfers fatty acids into peroxisomes, the cellular organelles where the fatty acids undergo β-oxidation.[8] A dysfunctional gene leads to the accumulation of very long chain fatty acids (VLCFA). The precise mechanisms through which high VLCFA concentrations cause the disease were still unknown as of 2005, but they do accumulate in the organs affected.
The incidence of X-linked adrenoleukodystrophy is at least 1 in 42,000 male births. [9]
Autosomal
Autosomal adrenoleukodystrophy has been associated with PEX1, PEX5, PEX10, PEX13, and PEX26.[10]
Treatment
While there is currently no cure for the disease, some dietary treatments, for example, a 4:1 mixture of glyceryl trioleate and glyceryl trierucate (Lorenzo's oil) in combination with a diet low in VLCSFA (very long chain saturated fatty acids), have been used with limited success, especially before disease symptoms appear. A 2005 study shows positive long-term results with this approach.[11] A 2007 report also appraises "Lorenzo's oil".[12] See also the Myelin Project. X-linked adrenoleukodystrophy has a very variable clinical course, even within a single family.[13] It is therefore not possible to determine if Lorenzo's oil is preventing progression of the disease in asymptomatic patients, or if these patients would have remained asymptomatic even without treatment. Current double blind placebo-controlled trials may be able to answer the questions regarding the effectiveness of treatment.
Hematopoietic stem cell transplantation (HSCT, including bone marrow transplant) is thought to be able to stop the progression of the disease in asymptomatic or mildly symptomatic boys who have a Loes score lower than 9 (an MRI measure of the severity of the disease), though outcomes are markedly poorer in symptomatic boys [14]. HSCT carries a risk of mortality and morbidity and is not recommended for patients whose symptoms are already severe. Umbilical cord blood stem cell transplant may provide an alternative for patients who do not have a matched related stem cell donor. Preliminary studies suggest that the outcome of cord blood stem cell transplant for ALD is particularly good in very young, presymptomatic patients [15].
Lovastatin is an anti-cholesterol drug that appears to have some effect in vitro, but not in mice with the animal model of adrenoleukodystrophy.[16] A clinical study of lovastatin showed encouraging biochemical changes, but no objective clinical improvement.[17] In a randomized, double-blind, placebo-controlled crossover trial, researchers for no effect of lovastatin on tissue levels of very long chain free fatty acids, and they recommended that it not be used in X-linked adrenoleukodystrophy.[18]
Currently, researchers at The Children's Hospital at the University of Minnesota, Dr. Charnas and Dr. Orchard, are investigating Mucomyst as an adjunct to bone marrow transplant, with some increase in survival time after transplant in 3 patients.[19]
According to a 1986 study, Oleic acid may lower the levels of very long chain free fatty acids in vitro. [20]
Research directions
Active clinical trials are currently in progress to see if proposed treatments are effective or not:[21]
- Glyceryl Trioleate (Lorenzo's oil) for Adrenomyelneuropathy.[22]
- Beta Interferon and Thalidomide[23] This study is closed.
- Combination of Glyceryl Trierucate and Glyceryl Trioleate (Lorenzo's Oil) in assymptomatic patients. [24]
- Hematopoietic stem cell transplantation[25]
Unproven New Therapies
In November 2009, Science published a report on a pilot study of two patients receiving gene therapy combined with blood-stem-cell therapy, and stating that the combination "may be a useful tool for treating" ALD.[26]
Presently, a boy with ALD is undergoing a new treatment in France. Professor Patrick Aubourg and Doctor Nathalie Cartier-Lacave are treating him with stem cells which have been altered by the introduction of inactivated AIDS virus cells. They have also successfully treated other children in this way. Says Nathalie Cartier-Lacave, “We have completely stabilised the evolution of this disease. These children are well, they go to school, they have a social life, a normal family life, and there’s no reason to think that this stabilisation isn’t permanent.” The technique was developed here in the Faculty of Pharmacy in Paris. First, stem cells are harvested from the patient’s soft bone tissue. Then a correcting gene is introduced using inactivated AIDS viral vectors. These are used because they are very effective at penetrating cells. Reinjected into the patients, the altered cells navigate straight to the brain where they permanently correct the gene deficiency which causes ALD. The therapy could also work for other more common diseases like hemophilia, thalassemia, Sickle Cell Anaemia, Parkinsons Disease and some cancers. If further research confirms these results, this could become standard treatment in the future – replacing bone transplants which are risky, invasive, and which require a compatible donor to be effective.[27]
Prognosis
Treatment is symptomatic. Progressive neurological degeneration makes the prognosis generally poor. Death occurs within one to ten years of presentation of symptoms. The use of Lorenzo's Oil, bone marrow transplant, and gene therapy is currently under investigation.
Lorenzo Odone
Lorenzo Michael Murphy Odone (May 29, 1978 – May 30, 2008) was probably the most famous patient with ALD. His parents Augusto and Michaela Odone, frustrated by the limited treatment available,[28] sparked the invention of "Lorenzo's oil", which is still being studied to see if it has therapeutic benefit in halting the destruction of the myelin sheathing of nerves caused by this disease. The quest for a treatment for Lorenzo was depicted in the 1992 film Lorenzo's Oil, and was the subject of the Phil Collins song "Lorenzo" (on his 1996 album Dance Into The Light).
References
- ↑ James, William D.; Berger, Timothy G.; et al. (2006). Andrews' Diseases of the Skin: clinical Dermatology. Saunders Elsevier. ISBN 0-7216-2921-0.
- ↑ Siemerling E, Creutzfeldt HG (1923). "Bronzekrankheit und sklerosierende Encephalomyelitis". Arch. Psychiat. Neurokrankh. 68: 217–44. doi:10.1007/BF01835678.
- ↑ O'Brien TJ, Gates PG, Byrne E (April 1996). "Symptomatic female heterozygotes for adrenoleukodystrophy: A report of two unrelated cases and review of the literature". Journal of Clinical Neuroscience : Official Journal of the Neurosurgical Society of Australasia 3 (2): 166–70. PMID 18638861. http://linkinghub.elsevier.com/retrieve/pii/S0967-5868(96)90012-0.
- ↑ Moser HW, Moser AB, Frayer KK, et al (October 1981). "Adrenoleukodystrophy: increased plasma content of saturated very long chain fatty acids". Neurology 31 (10): 1241–9. PMID 7202134.
- ↑ Moser HW, Raymond GV, Dubey P (Dec 2005). "Adrenoleukodystrophy: new approaches to a neurodegenerative disease". JAMA 294 (24): 3131–4. doi:10.1001/jama.294.24.3131. PMID 16380594.
- ↑ Bezman L, Moser AB, Raymond GV, Rinaldo P, Watkins PA, Smith KD, Kass NE, Moser HW (April 2001). "Adrenoleukodystrophy: incidence, new mutation rate, and results of extended family screening". Annals of Neurology 49 (4): 512–7. doi:10.1002/ana.101.
- ↑ Moser HW, Moser AB, Frayer KK, et al (Oct 1981). "Adrenoleukodystrophy: increased plasma content of saturated very long chain fatty acids". Neurology 31 (10): 1241–9. PMID 7202134.
- ↑ Mosser J, Douar AM, Sarde CO, et al (Feb 1993). "Putative X-linked adrenoleukodystrophy gene shares unexpected homology with ABC transporters". Nature 361 (6414): 726–30. doi:10.1038/361726a0. PMID 8441467.
- ↑ Bezman L, Moser HW (April 1998). "Incidence of X-linked adrenoleukodystrophy and the relative frequency of its phenotypes". American Journal of Medical Genetics 76 (5): 415–9. doi:10.1002/(SICI)1096-8628(19980413)76:5<415::AID-AJMG9>3.0.CO;2-L. PMID 9556301.
- ↑ Online 'Mendelian Inheritance in Man' (OMIM) ADRENOLEUKODYSTROPHY, AUTOSOMAL NEONATAL FORM -202370
- ↑ Moser, HW; Raymond GV, Lu S-E, Muenz LR, Moser AB, Xu J, Jones RO, Loes DJ, Melhem ER, Dubey P, Bezman L, Brereton NH, Odone A (2005-07). "Follow-up of 89 asymptomatic patients with adrenoleukodystrophy treated with Lorenzo's Oil.". Archives of Neurology 62 (7): p. 1073–80. doi:10.1001/archneur.62.7.1073. PMID 16009761.
- ↑ Moser HW, Moser AB, Hollandsworth K, Brereton NH, Raymond GV (Sep 2007). ""Lorenzo's oil" therapy for X-linked adrenoleukodystrophy: rationale and current assessment of efficacy". J. Mol. Neurosci. 33 (1): 105–13. doi:10.1007/s12031-007-0041-4. PMID 17901554.
- ↑ Online 'Mendelian Inheritance in Man' (OMIM) Adrenoleukodystrophy -300100
- ↑ Peters C, Charnas LR, Tan Y, Ziegler RS, Shapiro EG, DeFor T, Grewal SS, Orchard PJ, Abel SL, Goldman AI, Ramsay NK, Dusenbery KE, Loes DJ, Lockman LA, Kato S, Aubourg PR, Moser HW, Krivit W (2004). "Cerebral X-linked adrenoleukodystrophy: the international hematopoietic cell transplantation experience from 1982 to 1999". Blood 104 (3): 881–8. doi:10.1182/blood-2003-10-3402. PMID 15073029.
- ↑ Beam D, Poe MD, Provenzale JM, Szabolcs P, Martin PL, Prasad V, Parikh S, Driscoll T, Mukundan S, Kurtzberg J, Escolar ML (2007). "Outcomes of unrelated umbilical cord blood transplantation for X-linked adrenoleukodystrophy". Biol Blood Marrow Transplant 13 (6): 665–74. doi:10.1016/j.bbmt.2007.01.082. PMID 17531776.
- ↑ Yamada T, Shinnoh N, Taniwaki T, et al (September 2000). "Lovastatin does not correct the accumulation of very long-chain fatty acids in tissues of adrenoleukodystrophy protein-deficient mice". J. Inherit. Metab. Dis. 23 (6): 607–14. doi:10.1023/A:1005634130286. PMID 11032335. http://www.kluweronline.com/art.pdf?issn=0141-8955&volume=23&page=607.
- ↑ Pai GS, Khan M, Barbosa E, Key LL, Craver JR, Curé JK, Betros R, Singh I (April 2000). "Lovastatin therapy for X-linked adrenoleukodystrophy: clinical and biochemical observations on 12 patients". Molecular Genetics and Metabolism 69 (4): 312–22. doi:10.1006/mgme.2000.2977. PMID 10870849. http://linkinghub.elsevier.com/retrieve/pii/S1096719200929779.
- ↑ Engelen M, Ofman R, Dijkgraaf MG, et al. (January 2010). "Lovastatin in X-linked adrenoleukodystrophy". The New England Journal of Medicine 362 (3): 276–7. doi:10.1056/NEJMc0907735. PMID 20089986.Lovaststin should not be prescribed as therapy to lower levels of very-long-chain fatty acids in patients with X-ALD.
- ↑ Tolar J, Orchard PJ, Bjoraker KJ, Ziegler RS, Shapiro EG, Charnas L (Feb 2007). "N-acetyl-L-cysteine improves outcome of advanced cerebral adrenoleukodystrophy". Bone Marrow Transplant 39 (4): 211–5. doi:10.1038/sj.bmt.1705571. PMID 17290278.
- ↑ Rizzo WB, Watkins PA, Phillips MW, Cranin D, Campbell B, Avigan J (March 1986). "Adrenoleukodystrophy: oleic acid lowers fibroblast saturated C22-26 fatty acids". Neurology 36 (3): 357–61. PMID 3951702.
- ↑ clinicaltrials.gov/
- ↑ "A Phase III Trial of Lorenzo's Oil in Adrenomyeloneuropathy". http://www.clinicaltrials.gov/show/NCT00545597. Retrieved 2009-06-06.
- ↑ ClinicalTrials.gov NCT00004450
- ↑ "Study of Glyceryl Trierucate and glyceryl trioleate (Lorenzo's Oil) therapy in male children with adrenoleukodystrophy". http://www.clinicaltrials.gov/show/NCT00004418. Retrieved 2009-06-06.
- ↑ "HSCT for High Risk Inherited Inborn Errors". http://www.clinicaltrials.gov/show/NCT00383448. Retrieved 2009-06-06.
- ↑ "Gene Therapy Technique Slows Brain Disease ALD Featured In Movie 'Lorenzo's Oil'" at Science News site
- ↑ "Hematopoietic Stem Cell Gene Therapy with a Lentiviral Vector in X-Linked Adrenoleukodystrophy" at Science Mag site
- ↑ "About Lorenzo, his Parents, and Oumouri". The Myelin Project. http://www.myelin.org/aboutlorenzo.htm. Retrieved 2006-06-03.
External Links
- GeneReviews/NCBI/NIH/UW entry on Peroxisome Biogenesis Disorders, Zellweger Syndrome Spectrum
- GeneReviews/NCBI/NIH/UW entry on X-Linked Adrenoleukodystrophy
- Adrenoleukodystrophy at the Open Directory Project
- adrenoleukodystrophy at NINDS
- Images of ALD at USUHS
- Adrenoleukodystrophy at National Center for Biotechnology Information
- Information from ALDlife.org
|
|||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||